

Author: Felipe Paredes et al.
Date: January 2018
Poldip2 is an oxygen-sensitive protein that controls PDH and αKGDH lipoylation and activation to support metabolic adaptation in hypoxia and cancer
In this paper, the authors show that poldip2 governs the critical mechanism linking clp, ACSM1, and protein lipoylation; thus, regulating mitochondria function.
Lipoylation of two key enzymes in the TCA cycle, PDH and αKGDH, is a dynamically regulated process that is inhibited under hypoxia and in cancer cells, resulting in restricted mitochondria function. Poldip2, a ubiquitously expressed protein, regulates the lipoylation of both pyruvate and αKDH subunits via regulation of the clp-protease complex (CLPX ) and deregulation of the lipoate-activating enzyme required for lipoylation in mammalian cells, Ac-CoA synthetase (ACSM1). In Poldip2-defficient cells, repressed mitochondria function induces the stabilization of HIF-1α, thereby reprogramming cellular metabolism to resemble hypoxic/cancer cell adaption. Additionally, Poldip2 is down-regulated in hypoxic environments, further stabilizing HIF-1α while also inducing the expression of PDH kinase (PDK), consequently inhibiting lipoylation.
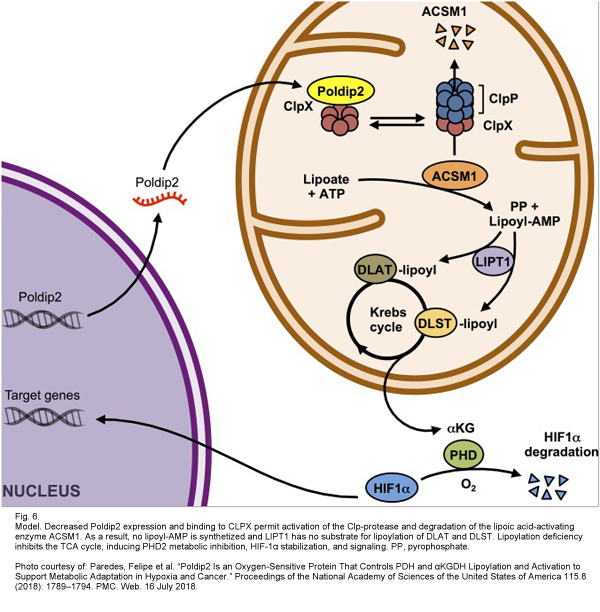 Poldip2 specifically binds to CLPX and sequesters it in normal cells; therefore, in poldip2-defficient cells, CLPX is capable of degrading ACSM1, impairing the lipoylation of PDH & αKGDH, limiting mitochondrial function, and stabilizing HIF-1α. This cascade was recapitulated in vitro by using the HypOxystation H35 to simulate a hypoxic environment so that the expression of poldip2 under oxygen tension could be analyzed. It was found that poldip2 was dramatically repressed under hypoxic conditions in a variety of cells. Additionally, it was proved that hypoxic conditions produced significantly reduced amounts of ASCM1 protein.
Poldip2 specifically binds to CLPX and sequesters it in normal cells; therefore, in poldip2-defficient cells, CLPX is capable of degrading ACSM1, impairing the lipoylation of PDH & αKGDH, limiting mitochondrial function, and stabilizing HIF-1α. This cascade was recapitulated in vitro by using the HypOxystation H35 to simulate a hypoxic environment so that the expression of poldip2 under oxygen tension could be analyzed. It was found that poldip2 was dramatically repressed under hypoxic conditions in a variety of cells. Additionally, it was proved that hypoxic conditions produced significantly reduced amounts of ASCM1 protein.
Interestingly, forced ACSM1 expression in poldip2-defficient cells was sufficient to rescue lipoylation levels and slow cancer growth rates, proving that the loss of ACSM1 is the primary mechanism by which poldip2-defficient cells inhibit mitochondrial function.
Implications of the aforementioned mechanism were analyzed in a cancer model. Impaired mitochondrial function and increased glycolysis create an advantageous environment for cancer cells to proliferate, survive, and become invasive in. It was hypothesized that cancer cells that express a highly glycolytic phenotype (OCR/ECAR = 2-5 pmol/mpH) are most likely poldip2-defficient. A specific sub-category of breast cancer known as triple negative breast cancer cells (TNBC) are known to be highly glycolytic, so the authors investigated whether poldip2-defficiency was involved. TNBC cell lines MDA-MB-231 and BT549 were found to have significantly lower poldip2 amounts compared to the non-TNBC cell line T47D. Again, a HypOxystation H35 was used to establish an accurate and reproducible hypoxic environment when analyzing the various cell lines.
Poldip2-defficiency is speculated to specifically impact the fate and availability of Ac-CoA. Since the TCA cycle is inhibited in poldip2-defficient cells, Ac-CoA could be redirected to biosynthetic pathways which may represent an advantage for cell proliferation or hypertrophy in tumor environments.
